
Dị tật bẩm sinh đường tiết niệu và thận ( CAKUT ) là một nhóm các bất thường về cấu trúc ảnh hưởng đến thận và đường tiết niệu. Ước tính có khoảng 4 đến 60 trường hợp trên 10.000 ca sinh trong toàn bộ dân số. Chẩn đoán CAKUT thường được thực hiện trong quá trình siêu âm thai định kỳ hoặc sau sinh ở trẻ sơ sinh có các dấu hiệu và triệu chứng lâm sàng liên quan. Tỷ lệ mắc CAKUT ở trẻ sinh non là 2% trong một nghiên cứu lớn gần đây được báo cáo trên nhóm 409.704 trẻ sơ sinh.
Dị tật phổ biến nhất là tắc nghẽn chỗ nối niệu quản – bể thận, ảnh hưởng đến khoảng 20% số người mắc bệnh. Trong số các loại CAKUT khác , có thận loạn sản đa nang, không có thận, loạn sản thận, thiểu sản thận, trào ngược bàng quang niệu quản, giãn niệu quản, hệ thống thu thập kép, niệu quản lạc chỗ và van niệu đạo sau (Hình 1).
Mặc dù đã có những tiến bộ đáng kể trong việc phát hiện và chẩn đoán CAKUT , nhưng nguyên nhân cơ bản của tình trạng này vẫn chưa được hiểu rõ hoàn toàn. Cần có thêm nghiên cứu để nâng cao hiểu biết của chúng ta về các yếu tố phôi thai học, di truyền và môi trường góp phần vào sự phát triển của CAKUT , cũng như để hiểu rõ hơn về nguyên nhân cơ bản và cải thiện các chiến lược phòng ngừa và điều trị.
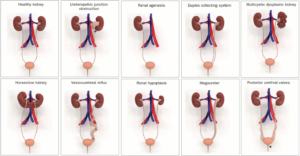
Hình 1. Mô hình 3D minh họa các dị tật bẩm sinh của thận và đường tiết niệu ( CAKUT ).
Phôi học
Sự phát triển của thận và đường tiết niệu ở thai nhi là một quá trình phức tạp bắt đầu từ những tuần đầu của thai kỳ và bao gồm sự hình thành và biệt hóa của nhiều cấu trúc. Quá trình này bắt đầu với sự hình thành các chồi niệu quản, phát sinh từ trung bì trung gian và tạo ra các ống góp và cuối cùng là thận. Thận cũng phát triển từ một cấu trúc gọi là metanephros, hình thành từ trung bì trong phôi thai.
Trong quá trình phát triển, các chồi niệu quản và thận sau phải kết nối và biệt hóa đúng cách để hình thành thận và đường tiết niệu chức năng. Sự gián đoạn trong quá trình này có thể dẫn đến một loạt các dị tật bẩm sinh.
CAKUT có thể được phân loại theo vị trí (thận, niệu quản, bàng quang hoặc niệu đạo), theo nguyên nhân (đột biến gen, tiếp xúc với môi trường, nhiễm trùng trong thai kỳ, v.v.), theo kiểu di truyền và theo mức độ nghiêm trọng (Bảng 1). Trong 10% đến 25% trường hợp, CAKUT là do rối loạn di truyền. CAKUT biểu hiện các triệu chứng kiểu hình phức tạp liên quan đến rối loạn di truyền (Hình 2,3). Các cá nhân có cùng bất thường về bộ gen có thể biểu hiện các dạng CAKUT khác nhau , và các bất thường kiểu hình tương tự có thể phát sinh do các rối loạn di truyền khác nhau, và có liên quan đến các bệnh ngoài thận.
Bảng 1. Các loại rối loạn CAKUT
| Quả thận | Hệ thống thu thập |
| Số: Thiếu thận bẩm sinh | Tắc nghẽn chỗ nối niệu quản – bể thận |
| Hình thái học: Giảm sản thận, loạn sản thận, MCDK | Trào ngược bàng quang niệu quản |
| Vị trí: Thận hình móng ngựa, lạc chỗ, thận vùng chậu | Giãn niệu quản |
| Hệ thống song song | |
| PUV |
Một số dị tật của nhu mô thận dẫn đến sự thất bại trong quá trình phát triển bình thường của các nephron, bao gồm loạn sản nang, loạn sản thận, không có thận, loạn sản ống thận và loạn sản ống thận.
Bệnh thận đa nang
Thuật ngữ bệnh thận đa nang thường được sử dụng để mô tả một bệnh trong đó có nhiều nang ở thận mà không kèm theo loạn sản.
Bệnh thận đa nang di truyền trội nhiễm sắc thể thường ( ADPKD ) và bệnh thận đa nang di truyền lặn nhiễm sắc thể thường ( ARPKD ) là hai loại bệnh thận đa nang phổ biến nhất. Bên cạnh kiểu di truyền, dù là di truyền trội hay lặn, ARPKD và ADPKD còn khác nhau về biểu hiện lâm sàng và tiên lượng. ADPKD là dạng PKD phổ biến nhất , và nó được gây ra bởi các đột biến trong gen PKD1 hoặc PKD2 .
Các gen liên quan đến sự phát triển của bệnh nang thận:
- PKD1 : Gen này có liên quan đến bệnh thận đa nang di truyền (ADPKD ). Đột biến gen PKD1 chịu trách nhiệm cho khoảng 85% trường hợp mắc ADPKD .
- PKD2 : Gen này cũng liên quan đến bệnh thận đa nang di truyền (ADPKD ). Đột biến gen PKD2 chịu trách nhiệm cho khoảng 15% trường hợp mắc ADPKD .
- HNF1β : Các đột biến trong gen này có liên quan đến bệnh thận đa nang di truyền trội trên nhiễm sắc thể thường loại 2 (ADPKD2).
- REN : Gen này có liên quan đến bệnh thận đa nang ở trẻ sơ sinh.
- MUC1 : Các đột biến trong gen này có liên quan đến bệnh thận nang tủy liên quan đến MUC1.
- INVS : Các đột biến trong gen này có liên quan đến bệnh thận nang tủy do teo ống thận.
- NPHP1 , NPHP3 , NPHP4 , NPHP5 , NPHP6 , NPHP7 , NPHP8 , NPHP9 : Các gen này có liên quan đến bệnh thận nang tủy do teo ống thận.
- BMP7 : Đột biến gen này có liên quan đến bệnh thận nang tủy do teo ống thận.
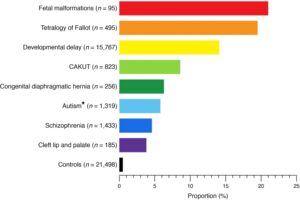
Hình 2. Tỷ lệ bệnh nhân mắc các rối loạn gen đã biết trong các kiểu hình phát triển khác nhau của con người và nhóm đối chứng khỏe mạnh.4
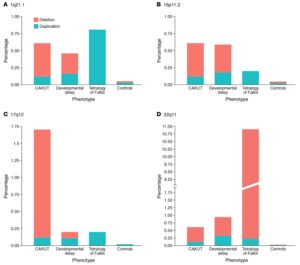
Hình 3. Sự khác biệt và tương đồng về tỷ lệ xuất hiện của bốn vị trí CNV thường gặp nhất ở bệnh nhân CAKUT .
MCDK
MCDK là bệnh nang bẩm sinh không di truyền, một dạng loạn sản thận gây ra nhiều nang không thông nhau, được ngăn cách bởi nhu mô loạn sản. Tỷ lệ mắc MCDK khoảng 1 trên 3.600 đến 1 trên 4.300 ca sinh sống. Nguyên nhân gây ra MCDK chưa được biết rõ, nhưng người ta cho rằng nó có thể liên quan đến sự hình thành bất thường của chồi niệu quản, tiếp xúc với các chất gây quái thai hoặc tắc nghẽn đường tiết niệu. MCDK thường được phát hiện thông qua siêu âm trước sinh. Vì thường không gây ra triệu chứng hoặc biến chứng, nên nó thường chỉ được phát hiện sau sinh nếu có một khối u đáng chú ý hoặc nếu được phát hiện tình cờ trong quá trình chụp chiếu cho một bệnh lý khác.
Một số gen có liên quan đến MCDK bao gồm:
- HNF1β : Gen này mã hóa một loại protein gọi là yếu tố hạt nhân tế bào gan 1 beta, đóng vai trò trong sự phát triển và chức năng của thận và các cơ quan khác. Đột biến trong HNF1β có thể gây ra một loạt các bất thường về thận, bao gồm cả MCDK .
- PAX2 : Gen này mã hóa một loại protein gọi là paired box 2, rất quan trọng cho sự phát triển của niệu quản, ống dẫn nước tiểu từ thận đến bàng quang. Đột biến ở gen PAX2 có thể gây ra bệnh MCDK và các dị tật thận khác.
- SIX1 : Gen này mã hóa một loại protein gọi là sine oculis homeobox 1, tham gia vào sự phát triển của nhiều cơ quan, bao gồm cả thận. Đột biến ở gen SIX1 có thể gây ra bệnh MCDK và các dị tật thận khác.
- WT1 : Gen này mã hóa một loại protein gọi là u Wilms 1, rất quan trọng cho sự phát triển của thận và các cơ quan khác. Đột biến ở gen WT1 có thể gây ra bệnh thận đa nang (MCDK) và các dị tật thận khác.
Hội chứng thận đa nang (MCDK ) thường được điều trị bảo tồn (tức là theo dõi) vì hầu hết các trường hợp không có biến chứng lâu dài. Bệnh nhân có bất thường ở thận đối bên có thể bị suy thận theo thời gian. Do đó, cần theo dõi lâu dài.
Thiếu thận bẩm sinh
So với chứng thiểu sản thận hai bên, thiểu sản thận một bên xảy ra ở một trong mỗi 1000 trường hợp mang thai và có tiên lượng tốt. Ước tính có khoảng 0,1% đến 0,3% trong số 1000 ca sinh bị ảnh hưởng bởi thiểu sản thận hai bên, thường dẫn đến tỷ lệ sống sót thấp. Một số kiểu di truyền đã được mô tả ở các gia đình mắc chứng thiểu sản thận, có thể xảy ra như một phát hiện riêng lẻ hoặc là một phần của hội chứng.
Chứng thiểu sản thận một bên có thể do đột biến ở một số gen, bao gồm ATRX , RET , BMP4 , FRAS1 , FREM1 , GRIP1 , GLI3 , UPK3A , và nhiều gen khác. Ngoài ra, chứng thiểu sản thận hai bên có thể do đột biến ở các gen RET , FGF20 , ITGA8 . Các gen liên quan được liệt kê trong Bảng 2 .
Loạn sản thận
Loạn sản thận là kết quả của sự ngừng phát triển trong ba tháng đầu đời của thai kỳ ở mầm thận. Tuy nhiên, nguyên nhân gây ra loạn sản thận vẫn chưa được biết rõ; bất thường mạch máu trong tử cung cũng như rối loạn phát triển di truyền là hai cơ chế có thể xảy ra. Loạn sản thận có thể là một phát hiện đơn độc hoặc là một phần của hội chứng di truyền liên quan đến dị tật đường tiết niệu. Nghiên cứu ESCAPE đã phân tích các gen phát triển ( HNF1β , PAX2 , EYA1 , SIX1 và SALL1 ) liên quan đến loạn sản thận ở một nhóm lớn trẻ em mắc loạn sản thận. Theo nghiên cứu này, đột biến PAX2 được chẩn đoán ở 15% bệnh nhân mắc bệnh lý thận, và những người có đột biến hoặc mất đoạn gen ở các gen lớn cho thấy kiểu hình thận khác nhau. Ngoài ra, 22% tổng số bệnh nhân mắc loạn sản thận dạng nang có đột biến HNF1, cho thấy rằng những người mắc loạn sản thận dạng nang nên được sàng lọc đột biến HNF1 . Kết quả siêu âm thận cho thấy kích thước thận giảm và mất sự phân biệt vỏ tủy thận là dấu hiệu lâm sàng của chứng thiểu sản thận. Một số bệnh nhân bị thiểu sản thận có thể tiến triển đến bệnh thận giai đoạn cuối theo thời gian. Hiện chưa có phương pháp điều trị đặc hiệu cho chứng thiểu sản thận. Việc theo dõi và quản lý chức năng thận trong nhiều năm là rất quan trọng. Đối với những bệnh nhân tiến triển đến bệnh thận giai đoạn cuối, ghép thận là lựa chọn ưu tiên.
Hẹp khúc nối niệu quản – bể thận
Nguyên nhân bệnh lý phổ biến nhất gây giãn thận ở trẻ sơ sinh là tắc nghẽn phần trên của niệu quản. Tỷ lệ mắc bệnh chung là 1:1.500, với tỷ lệ 2:1 giữa trẻ sơ sinh nam và nữ. Sự tắc nghẽn ở chỗ nối niệu quản – bể thận thường do hẹp bẩm sinh hoặc, ít phổ biến hơn, do một mạch máu bắt chéo gây chèn ép từ bên ngoài ở phần trên của niệu quản. Một cơ chế có thể là do sự gián đoạn trong quá trình phát triển phôi thai ở niệu quản gần, làm thay đổi sự phát triển của cơ vòng, bằng cách làm thay đổi các sợi collagen và thành phần giữa và xung quanh các tế bào cơ. Một số đột biến gen đơn lẻ có liên quan đến UPJO , bao gồm Id2 , PAX2 , EYA , AGTR2 , BMP4 , SOX17 , CHD1L và DSTYK . Id2 và Adamts1 được báo cáo là tạo ra kiểu hình tương tự như kiểu hình UPJO ở người . Chuột thiếu Id2 cũng biểu hiện vị trí cắm niệu quản vào bể thận cao.
Trào ngược bàng quang niệu quản
Một số gen liên quan đến sự phát triển của bệnh trào ngược bàng quang niệu quản (VUR) đã được xác định. Một số gen này tham gia vào sự phát triển và chức năng của cơ trơn niệu quản. Các gen khác tham gia vào sự phát triển và chức năng của bàng quang. Tỷ lệ mắc VUR cao ở các cặp song sinh, anh chị em ruột và con cái cho thấy bệnh này có cơ sở di truyền. Dựa trên kết quả nghiên cứu di truyền ở nhiều quần thể khác nhau, người ta đã xác định rằng VUR là một bệnh có tính chất di truyền không đồng nhất. Một nghiên cứu liên kết và hiệp hội trên toàn bộ hệ gen đã liên kết VUR nguyên phát không kèm hội chứng với vùng 10q26 của bộ gen.20,21Các gen khác có liên quan đến sự phát triển của VUR bao gồm gen FOXC1 , gen MYH11 và gen ACTN4 . Những gen này đóng vai trò khác nhau trong sự phát triển và chức năng của đường tiết niệu. Một nghiên cứu liên kết toàn bộ hệ gen sử dụng phân tích biến thể số lượng bản sao lớn nhất và nghiên cứu liên kết toàn bộ hệ gen về bệnh trào ngược bàng quang niệu quản (VUR) đang dẫn đến việc xác định năm vị trí có thể liên quan đến VUR , bao gồm WDPCP , OTX1 , BMP5 , WDPCP và WNT5 với tác động lớn. WNT5 có thể đóng vai trò trong sự phát triển của hệ tiết niệu sinh dục. Một nghiên cứu liên kết toàn bộ hệ gen sử dụng phân tích biến thể số lượng bản sao lớn nhất và nghiên cứu liên kết toàn bộ hệ gen về bệnh VUR đang dẫn đến việc xác định năm vị trí có thể liên quan đến VUR , bao gồm WDPCP , OTX1 , BMP5 , WDPCP và WNT5 với tác động lớn. WNT5 có thể đóng vai trò trong sự phát triển của hệ tiết niệu sinh dục.
Van niệu đạo sau
Hẹp van niệu đạo sau là một dị tật bẩm sinh đe dọa tính mạng của đường tiết niệu, thường được phát hiện trong giai đoạn phát triển sơ sinh và, mặc dù được điều trị tối ưu, vẫn dẫn đến tỷ lệ suy thận cao. Ước tính hẹp van niệu đạo sau xảy ra ở 1 trong 7.000–8.000 ca sinh sống.
Có nhiều báo cáo chỉ ra rằng hẹp niệu đạo bẩm sinh (PUV) là nguyên nhân hàng đầu gây bệnh thận mãn tính ở trẻ sơ sinh nam, và suy thận giai đoạn cuối (ESRD) có thể xảy ra ở 5% đến 64% trường hợp. Một đánh giá có hệ thống cho thấy bệnh thận mãn tính ( CKD ) và bệnh thận giai đoạn cuối (ESKD) có liên quan đến bệnh nhân PUV với nguy cơ lần lượt lên tới 32% và 20%.
Cho đến nay, chỉ có một vài gen được xác định có liên quan đến van niệu đạo sau. Hội chứng Prune Belly chỉ được liên kết với gen HNF1B trong 3% trường hợp được ghi nhận, và hầu hết những trường hợp được chẩn đoán đều không có nguyên nhân di truyền. Trong số các dị tật CAKUT liên quan đến hội chứng Down (Trisomy 21), có van niệu đạo sau, giãn bể thận và giãn niệu quản.
Bảng 2. Tóm tắt các gen liên quan đến sự phát triển của CAKUT.
| Rối loạn | Gen |
| Thiếu sản/loạn sản thận | ACE , AGT , ATRX , CHD7 , ESCO2 , EYA1 , SIX1 , FRAS1 , FREM2 , GATA3 , GLI3 , GPC3 , GRIP1 , HNF1β , JAG1 , KAL1 , LRP4 , MKS1–4 , NIPBL , PAX2 , PBX1 , REN , SALL1 , SIX5 |
| Thiếu thận bẩm sinh | ATRX , BMP4 , CBP/EP300 , CHD7 , ESCO2 , FGF20 , FRAS1 , FREM2 , GATA3 , GRIP1 , GLI3 , HNF1β , ITGA8 , JAG1 , KAL1 , KAL2 , RET , SALL1 , VANGL1 |
| Loạn sản dạng nang | CBP/EP300 , DHCR7 , EVC , EVC2 , FRAS1 , FREM2 , GPC-3 , HNF1β , JAG1 , KAL1 , KAL2 , MKS1–4 , NS1 , PEX , PAX2 , WT1 |
| Hệ thống song công | NS1 |
| Thận hình móng ngựa/thận lạc chỗ | GLI3 , NIPBL , VANGL1 |
| Ứ nước thận, UPJ | ACE , Adamts-1 , AGT , ATRX , BMP4 , CHD1L , CHD7 , DHCR7 , DSTYK , ESCO2 , EYA , FRAS1 , FREM2 , GLI3 , HSPG2 , JAG1 , Id2 , NS1 , PAX2 , PEX , RET , SOX17 , VANGL1 |
| Hidroureter, Giãn niệu quản | CHD7 , GLI3 |
| VUR | ATRX , DHCR7 , EYA1 , FOX1 , GATA3 , HOXA13 , HPSE2 , JAG1 , KAL1 , KAL2 , MYH11 , NIPBL , PAX2 , SALL1 , SIX1 , SIX5 |
| PUV | BNC2 , SALL1 |
Tầm quan trọng lâm sàng và ý nghĩa tương lai của kiến thức về gen
Khi lĩnh vực đánh giá di truyền tiếp tục phát triển, có khả năng chúng ta sẽ ngày càng có khả năng xác định được các nguyên nhân di truyền của CAKUT và dự đoán được khả năng phát triển các bệnh lý này. Điều này có thể dẫn đến chẩn đoán và điều trị sớm hơn, từ đó cải thiện kết quả điều trị cho những người bị ảnh hưởng.
Cần lưu ý rằng xét nghiệm di truyền không phải lúc nào cũng cần thiết để chẩn đoán CAKUT . Chẩn đoán xác định thường được đưa ra dựa trên sự kết hợp giữa biểu hiện lâm sàng, tiền sử bệnh và các xét nghiệm hình ảnh. Xét nghiệm di truyền đối với CAKUT thường được thực hiện tại các trung tâm chuyên khoa dưới sự giám sát của bác sĩ di truyền lâm sàng, vì việc giải thích kết quả có thể phức tạp và có thể ảnh hưởng đến các quyết định về sinh sản. Một số phòng thí nghiệm (PreventionGenetics, Invitae, Blueprint Genetics, v.v.) cung cấp xét nghiệm di truyền cho một số rối loạn CAKUT khác nhau . Gói xét nghiệm của họ đối với CAKUT bao gồm xét nghiệm di truyền cho nhiều gen liên quan đến rối loạn CAKUT , bao gồm HNF1B , NPHP1 và UPK1B . Xét nghiệm di truyền đối với các rối loạn này có thể giúp xác nhận chẩn đoán, cung cấp thông tin về mức độ nghiêm trọng của rối loạn và hỗ trợ tư vấn di truyền và kế hoạch hóa gia đình. Cần lưu ý rằng các công ty và phòng thí nghiệm xét nghiệm di truyền khác cũng có thể có CAKUT trong gói xét nghiệm của họ. Vì CAKUT là một nhóm các rối loạn do các đột biến gen khác nhau gây ra, nên xét nghiệm gen chẩn đoán CAKUT ở một số công ty có thể không bao gồm tất cả các nguyên nhân di truyền của CAKUT , và các phòng thí nghiệm khác nhau có thể có danh sách gen được xét nghiệm khác nhau.
Hiểu biết đầy đủ về các thành phần di truyền của CAKUT là điều cần thiết để phát triển các chiến lược xét nghiệm di truyền chính xác. Ngoài ra, sự hiểu biết này sẽ hỗ trợ việc đưa ra quyết định lâm sàng trong xét nghiệm di truyền. Bao gồm các kết quả lâm sàng quan trọng và chức năng thận, các nghiên cứu theo dõi dài hạn là cần thiết để điều tra tác động lâu dài của việc chẩn đoán và quản lý sớm CAKUT .
Kết luận và tóm tắt
Dị tật bẩm sinh đường tiết niệu và thận ( CAKUT ) là những bất thường về cấu trúc ảnh hưởng đến thận và đường tiết niệu. Ước tính có khoảng 4-60 trường hợp trên 10.000 ca sinh. Chẩn đoán CAKUT thường được thực hiện trong quá trình siêu âm trước sinh hoặc sau khi sinh ở trẻ sơ sinh có các dấu hiệu và triệu chứng lâm sàng liên quan. Dị tật phổ biến nhất là tắc nghẽn chỗ nối niệu quản – bể thận, ảnh hưởng đến khoảng 20% người mắc bệnh. Sự phát triển của thận và đường tiết niệu bao gồm sự hình thành và biệt hóa của một số cấu trúc, bao gồm chồi niệu quản và thận nguyên thủy, phải kết nối đúng cách để tạo thành thận và đường tiết niệu hoạt động. Sự gián đoạn trong quá trình này có thể dẫn đến CAKUT . Có nhiều loại rối loạn CAKUT khác nhau, bao gồm những rối loạn ảnh hưởng đến số lượng, hình thái và vị trí của thận, và những rối loạn ảnh hưởng đến hệ thống thu thập nước tiểu. Trong 10-25% trường hợp, CAKUT là do rối loạn di truyền. Một số gen đã được xác định có liên quan đến sự phát triển của các bệnh lý khác nhau liên quan đến thận. Các nghiên cứu đang được tiến hành để nâng cao hiểu biết của chúng ta về nguyên nhân gây ra CAKUT và để phát triển các chiến lược phòng ngừa và điều trị tốt hơn. Mặc dù còn nhiều hạn chế và khó khăn hiện tại, chẩn đoán di truyền phân tử có thể mang lại hy vọng cải thiện việc quản lý lâm sàng cho bệnh nhân mắc CAKUT trong tương lai.
Điểm chính
- Dị tật bẩm sinh đường tiết niệu và thận ( CAKUT ) là một nhóm các bất thường về cấu trúc ảnh hưởng đến thận và đường tiết niệu, với ước tính khoảng 4 đến 60 trường hợp trên 10.000 ca sinh trong toàn bộ dân số.
- Sự phát triển của thận và đường tiết niệu ở thai nhi là một quá trình phức tạp bắt đầu từ những tuần đầu của thai kỳ và bao gồm sự hình thành và biệt hóa của nhiều cấu trúc. Sự gián đoạn trong quá trình này có thể dẫn đến một loạt các dị tật bẩm sinh.
- Trong một nghiên cứu quy mô lớn gần đây với 409.704 trẻ sơ sinh,tỷ lệ mắc CAKUT ở trẻ sinh non là 2%.
- Dạng dị tật phổ biến nhất là tắc nghẽn chỗ nối niệu quản – bể thận, ảnh hưởng đến khoảng 20% số người mắc bệ
- Trong 10-25% trường hợp, CAKUT là do rối loạn di truyề
- Việc đánh giá di truyền đang ngày càng tiến bộ, giúp tăng khả năng xác định nguyên nhân di truyền của CAKUT và dự đoán khả năng mắc phải tình trạng này.
- Xét nghiệm di truyền không phải lúc nào cũng cần thiết để chẩn đoán CAKUT ; việc chẩn đoán thường dựa trên sự kết hợp giữa biểu hiện lâm sàng, tiền sử bệnh và các xét nghiệm hình ả
- Hiểu biết đầy đủ về các thành phần di truyền của CAKUT là điều cần thiết để phát triển các chiến lược xét nghiệm di truyền chính xác và hướng dẫn việc ra quyết định lâm sàng trong xét nghiệm di truyề
- Cần tiến hành các nghiên cứu theo chiều dọc để điều tra tác động lâu dài của việc chẩn đoán và quản lý sớm bệnh CAKUT .
